
![]()
🚀 neuroSCC Bridging Simultaneous Confidence
Corridors and PET Neuroimaging. This package facilitates
structured processing of PET neuroimaging data for the estimation of
Simultaneous Confidence Corridors (SCCs). It integrates neuroimaging and
statistical methodologies to:
The package bridges established neuroimaging
tools (oro.nifti) with advanced statistical
methods (ImageSCC), supporting
one-group, two-group, and single-patient vs. group
comparisons.
📌 Developed as part of the Ph.D. thesis: “Development of statistical methods for neuroimage data analysis towards early diagnosis of neurodegenerative diseases”, by Juan A. Arias at University of Santiago de Compostela (Spain).
💡 This work was partially supported by an internship grant awarded at the 6th Conference of the Spanish National Biostatistics Network (BIOSTATNET) in 2025, as a prize for best poster presentation and young researcher trajectory.
neuroSCC?PET neuroimaging data is complex, requiring careful
processing and statistical validation.
neuroSCC is designed to:
✔ Automate Preprocessing: Load, clean, and structure
PET data 📂
✔ Standardize Analysis: Convert images into
FDA-compatible formats 🔬
✔ Evaluate SCC Estimations: Identify
significant regions with confidence 🎯
✔ Enable Method Comparisons: SCC vs SPM
performance evaluation 📊
It is particularly suited for: - Clinical neuroimaging research (Alzheimer’s disease, neurodegeneration). - Statistical validation of imaging methods. - Comparisons between SCC and other statistical approaches.
# Install the latest stable release (Future)
remotes::install_github("iguanamarina/neuroSCC@1a91f8e")
library(neuroSCC)# Install the latest development version
remotes::install_github("iguanamarina/neuroSCC")
library(neuroSCC)# Once available on CRAN
install.packages("neuroSCC")
library(neuroSCC)neuroCleaner() reads NIFTI neuroimaging
files, extracts voxel-wise data, and
structures it into a tidy data frame.
It is the first preprocessing step, ensuring that PET
images are cleaned and formatted for further analysis. It also
integrates demographic data when available.
# Load a sample Control NIfTI file
niftiFile <- system.file("extdata", "syntheticControl1.nii.gz", package = "neuroSCC")
# Example Without demographic data
petData <- neuroCleaner(niftiFile)
petData[sample(nrow(petData), 10), ] # Show 10 random voxelsdatabaseCreator() scans a directory for PET
image files, processes each with neuroCleaner(),
and compiles them into a structured data frame.
This function is critical for batch analysis, preparing
data for group-level SCC comparisons.
#' @examples
#' # NOTE: To keep runtime below CRAN limits, this example processes only 1 subject.
#' # You can expand the pattern to include all subjects for real use.
#'
#' # Example: Create a database from a single synthetic PET image (control group)
#' controlPattern <- "^syntheticControl1\\.nii\\.gz$"
#' databaseControls <- databaseCreator(pattern = controlPattern, control = TRUE, quiet = TRUE)
#'
#' head(databaseControls)getDimensions() extracts the spatial
dimensions of a neuroimaging file, returning the number of
voxels in the x, y, and z axes.
This ensures proper alignment of neuroimaging data before further
processing.
# Extract spatial dimensions of a PET scan
niftiFile <- system.file("extdata", "syntheticControl1.nii.gz", package = "neuroSCC")
dims <- getDimensions(niftiFile)
print(dims)matrixCreator() transforms PET imaging data into
a matrix format for functional data analysis.
Each row represents a subject’s PET data, formatted to align with FDA
methodologies.
# NOTE: To keep example runtime short, only one synthetic PET file is used.
# For full analysis, expand the filename pattern accordingly.
# Step 1: Generate a database for a single subject
controlPattern <- "^syntheticControl1\\.nii\\.gz$"
databaseControls <- databaseCreator(pattern = controlPattern, control = TRUE, quiet = TRUE)
# Step 2: Convert the database into a matrix format
matrixControls <- matrixCreator(databaseControls, paramZ = 35, quiet = TRUE)
# Display dimensions of the matrix
dim(matrixControls)meanNormalization() performs row-wise mean
normalization, adjusting intensity values across
subjects.
This removes global intensity differences, making datasets comparable in
Functional Data Analysis (FDA).
# Generate a minimal database and create a matrix (1 control subject)
dataDir <- system.file("extdata", package = "neuroSCC")
controlPattern <- "^syntheticControl1\\.nii\\.gz$"
databaseControls <- databaseCreator(pattern = controlPattern,
control = TRUE,
quiet = TRUE)
matrixControls <- matrixCreator(databaseControls, paramZ = 35, quiet = TRUE)
# Normalize the matrix (with diagnostics)
normalizationResult <- meanNormalization(matrixControls,
returnDetails = TRUE,
quiet = FALSE)neuroContour() extracts region boundaries
(contours) from neuroimaging data.
It is particularly useful for defining masks or Regions of
Interest (ROIs) before SCC computation.
# Get the file path for a sample NIfTI file
niftiFile <- system.file("extdata", "syntheticControl1.nii.gz", package = "neuroSCC")
# Extract contours at level 0
contours <- neuroContour(niftiFile, paramZ = 35, levels = 0, plotResult = TRUE)
# Display the extracted contour coordinates
if (length(contours) > 0) {
head(contours[[1]]) # Show first few points of the main contour
}getPoints() identifies regions with significant
differences from an SCC computation.
After ImageSCC::scc.image() computes SCCs,
getPoints() extracts coordinates where group
differences exceed confidence boundaries.
# Load precomputed SCC example
data("SCCcomp", package = "neuroSCC")
# Extract significant SCC points
significantPoints <- getPoints(SCCcomp)
# Show first extracted points (interpretation depends on SCC computation, see description)
head(significantPoints$positivePoints) # Regions where Pathological is hypoactive vs. Control
head(significantPoints$negativePoints) # Regions where Pathological is hyperactive vs. ControlgetSPMbinary() extracts significant
points from an SPM-generated binary NIfTI
file.
It returns voxel coordinates where SPM detected significant
differences, making it comparable to SCC results.
# Load a sample binary NIfTI file (SPM result)
niftiFile <- system.file("extdata", "binary.nii", package = "neuroSCC")
detectedSPM <- getSPMbinary(niftiFile, paramZ = 35)
# Show detected points
head(detectedSPM)processROIs() processes Regions of Interest
(ROIs) from neuroimaging files.
It extracts voxel coordinates for predefined hypoactive
regions, structuring them for SCC analysis.
# Load and process a sample ROI NIfTI file (console output)
roiFile <- system.file("extdata", "ROIsample_Region2_18.nii.gz", package = "neuroSCC")
processedROI <- processROIs(roiFile, region = "Region2", number = "18", save = FALSE)
head(processedROI)generatePoissonClones() creates synthetic clones
of PET neuroimaging data by adding Poisson-distributed noise.
This function is essential for 1 vs. Group SCC
analyses, where a single subject’s data needs to be expanded to
allow for valid statistical inference.
# Load example input matrix for Poisson cloning
data("generatePoissonClonesExample", package = "neuroSCC")
# Select 10 random voxel positions for display
set.seed(123)
sampledCols <- sample(ncol(generatePoissonClonesExample), 10)
# Generate 1 synthetic clone
clones <- generatePoissonClones(generatePoissonClonesExample, numClones = 1, lambdaFactor = 0.25)
# Show voxel intensity values after cloning
clones[, sampledCols]calculateMetrics() assesses the accuracy of
SCC-detected significant points by comparing them to
known true ROI regions. It computes
Sensitivity, Specificity, PPV, and NPV, allowing for a
quantitative evaluation of SCC performance.
data("calculateMetricsExample", package = "neuroSCC")
# Evaluate SCC and SPM detection performance
with(calculateMetricsExample, {
metricsSCC <- calculateMetrics(detectedSCC, trueROI, totalCoords, "Region2_SCC")
metricsSPM <- calculateMetrics(detectedSPM, trueROI, totalCoords, "Region2_SPM")
print(metricsSCC)
print(metricsSPM)
})A full walkthrough of using neuroSCC from start to
finish is available in the vignettes:
📌 Landing
Vignette
Covers data loading, matrix creation, and
triangulations.
📌 Two-group
SCC Estimation & Comparison
Computes SCCs for the differences between two groups and identifies
voxels outside of estimated confidence intervals.
📌 1vsGroup
SCC Estimation & Comparison
Compares an individual patient to a control group using SCCs and
identifies voxels outside of estimated confidence
intervals.
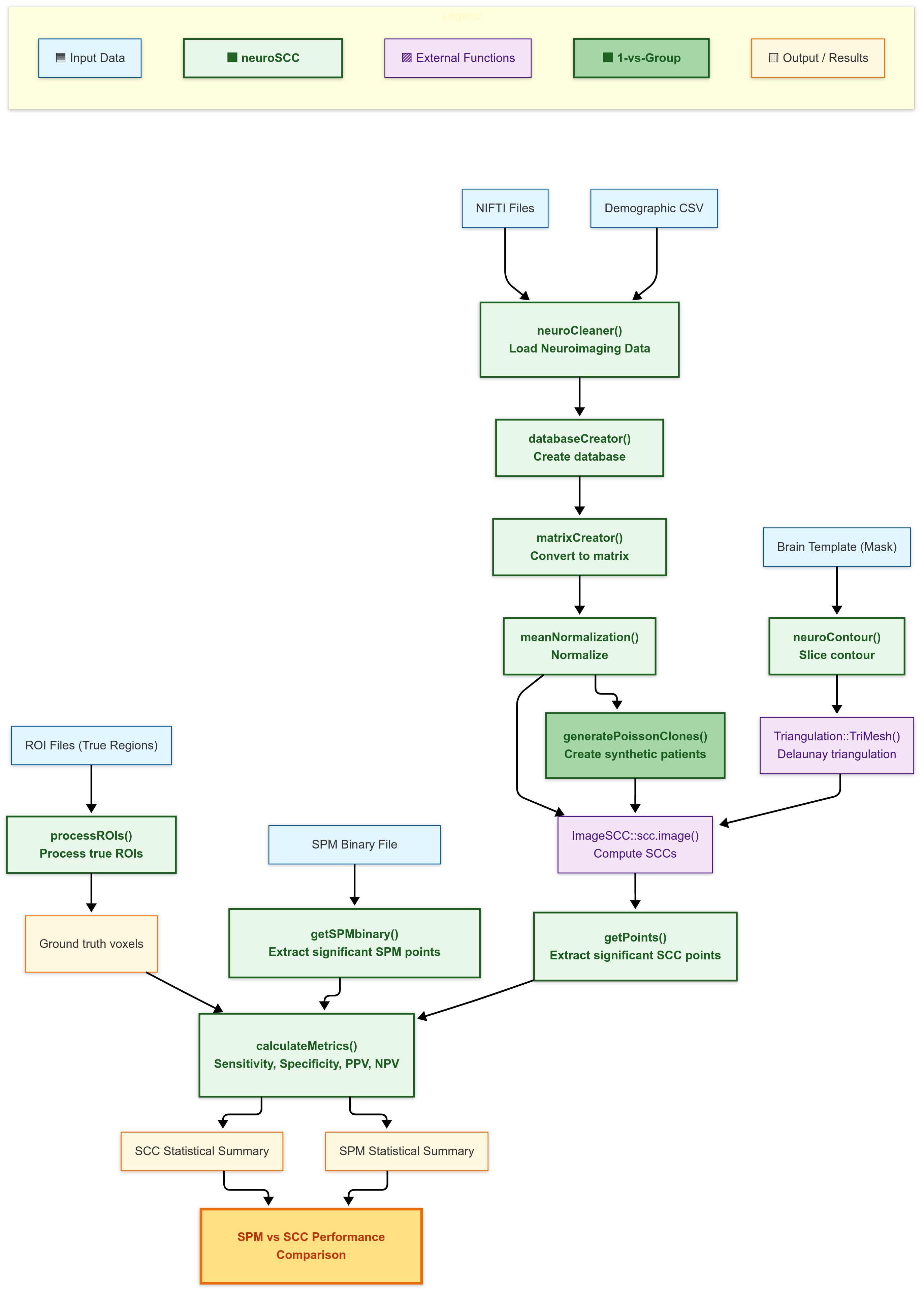
A complete visual overview of how neuroSCC functions
interact with data, the objects they return, and more, can be found in
the Visual Workflow:
Wang, Y., Wang, G., Wang, L., & Ogden, R.T. (2020). Simultaneous Confidence Corridors for Mean Functions in Functional Data Analysis of Imaging Data. Biometrics, 76(2), 427-437. doi:10.1111/biom.13156
Arias-López, J. A., Cadarso-Suárez, C., & Aguiar-Fernández, P. (2021). Computational Issues in the Application of Functional Data Analysis to Imaging Data. In International Conference on Computational Science and Its Applications (pp. 630–638). Springer International Publishing, Cham. doi:10.1007/978-3-030-86960-1_46
Arias-López, J. A., Cadarso-Suárez, C., & Aguiar-Fernández, P. (2022). Functional Data Analysis for Imaging Mean Function Estimation: Computing Times and Parameter Selection. Computers, 11(6), 91. MDPI. doi:10.3390/computers11060091
Ph.D. Thesis: Arias-López, J. A. (Under development). Development of Statistical Methods for Neuroimage Data Analysis Towards Early Diagnosis of Neurodegenerative Diseases. University of Santiago de Compostela.
We welcome contributions, feedback, and issue
reports from the community! If you would like to help improve
neuroSCC, here’s how you can get involved:
If you encounter a bug, incorrect result, or any unexpected behavior, please:
We are always looking to improve neuroSCC. If you have a
suggestion for a new feature or an enhancement,
please:
We love contributions! To submit a pull request (PR):
Fork the repository on GitHub.
Clone your fork to your local machine:
git clone https://github.com/YOUR_USERNAME/neuroSCC.git
cd neuroSCCCreate a new branch for your feature or fix:
git checkout -b feature-new-functionalityMake your changes and commit them:
git add .
git commit -m "Added new functionality XYZ"Push your changes to your fork:
git push origin feature-new-functionalitySubmit a pull request (PR) from your forked
repository to the main neuroSCC repository.
Before submitting, please:
✔ Ensure your code follows the package style
guidelines.
✔ Add documentation for any new functions or
features.
✔ Run devtools::check() to verify that all
package tests pass.
For questions not related to bugs or feature requests, feel free
to:
📬 Email the maintainer:
juanantonio.arias.lopez@usc.es
💬 Join the discussion on GitHub
Discussions